سندرم هانتر
سندرم Hunter یا Mucopolysaccharidosis 2 (یا MPS 2 ) یک بیماری ذخیره سازی لیزوزومی ناشی از آنزیم معیوب (یا عدم وجود)، iduronate-2-sulfatase (I2S) است.این سندرم دارایزمینه ی ارثی است که توسط X کنترل می شود.
رئوس مطالب
- 1 علائم و نشانه ها
- 2 پاتوفیزیولوژی
- 2.1 ژنتیک
- 2.2 بیوشیمی
- 3 تشخیص
- 4 درمان
- 4.1 درمان تسکین دهنده
- 4.2 پیوند مغز استخوان
- 4.3 جایگزینی آنزیم
- 5 اپیدمیولوژی
- 6 تاریخچه
علائم و نشانه ها
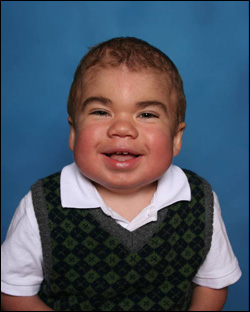
علائم سندرم Hunter (MPS II) معمولا در هنگام تولد مشخص نیست، اما معمولا بعد از سال اول قابل تشخیص است. اغلب اولین نشانه های سندرم هانتر ممکن است شامل فتق های شکمی ، عفونت گوش ، آبریزش بینی و سرماخوردگی باشد. از آنجایی که این علائم در میان تمام نوزادان شایع است، احتمالا به پزشک مراجعه نمیکنند تا تشخیص سندرم هانتر را صورت گیرد. با افزایش تولید گلیکواسامینوگرین ها (GAG) در سراسر سلول های بدن، علائم سندرم هانتر بیشتر از پیش قابل مشاهده است. علایم فیزیکی بسیاری از کودکان مبتلا به سندرم هانتر شامل یک برآمدگی متمایز در ویژگی های صورت خود، از جمله پیشانی برجسته، بینی با برآمدگی و زبان بزرگ است. آنها همچنین ممکن است سر بزرگ و همچنین شکم بزرگ داشته باشند. بسیاری از آن ها عفونت های مکرر گوش و دستگاه تنفسی را با خود به همراه دارند .
ذخیره و نگهداری GAG در سلول ها می تواند منجر به ارگان هایی که از راه های مهم تحت تاثیر قرار می گیرند. ضخیم شدن دریچه های قلب همراه با دیواره های قلب می تواند منجر به کاهش عملکرد قلب شود. دیواره های راه هوایی نیز ممکن است ضخیم شود، که در زمان خواب تنفس پر سر و صدا را باعث می شود. افراد مبتلا به سندرم هانتر همچنین ممکن است ظرفیت ریه شان محدود شود با توجه به اینکه کبد و طحال در طول زمان بزرگتر می شوند، شکم ممکن است دچار اختلال شود ، و در هم فرو رفتگی بیشتری ایجاد می کند.
همه مفاصل بزرگ (از جمله مچ دست ، آرنج ، شانه ، باسن و زانو ) ممکن است تحت تاثیر سندرم هانتر قرار بگیرند، که منجر به سفتی مفصلی و حرکت محدود می شود. دخالت مداوم انگشت و انگشت شست باعث کاهش توانایی انتخاب اشیاء کوچک می شود. اثرات بر روی مفاصل دیگر، مانند مفصل ران و زانو، می تواند باعث ایجاد سختی و مشکل در راه رفتن بشود. خود استخوان ها ممکن است تحت تأثیر قرار بگیرند و در نتیجه کوتاه شدن قد. علاوه بر این، ضایعات پوستی ضخیم، عاج رنگی ممکن است بر روی سینه ها، پاها و پشتی برخی افراد مبتلا به سندرم هانتر پیدا شود. وجود یا عدم وجود ضایعات پوستی در پیش بینی بالینی در سندرم هانتر مفید نیست. در نهایت، ذخیره سازی GAG در مغز می تواند منجر به تأخیر در رشد مغز با عقب ماندگی ذهنی و از دست دادن عملکرد پیشرفته شود. میزان و درجه پیشرفت برای هر فرد با سندرم هانتر متفاوت است.
گرچه سندرم هانتر با طیف گسترده ای از شدت بالینی همراه است، می توان دو فرم اصلی را تشخیص داد:
شدید و ملایم / ضعیف.
تفاوت بین اشکال شدید و ضعیف به طور عمده به توسعه مترقی عصبی شدن در فرم شدید است. مهم است که توجه داشته باشید، هرچند که اصطلاحات "ضعیف" یا "خفیف" توسط پزشکان در مقایسه با افراد مبتلا به سندرم هانتر مورد استفاده قرار می گیرند، اثرات بیماری خفیف بسیار جدی است. بین دو شکل اصلی بیماری وشکل درون آنها، دو مورد از مهمترین زمینه هاای قابل توجه مربوط به میزان عقب ماندگی ذهنی و طول عمر مورد انتظار است . برخی از افراد مبتلا به سندرم هانتر هیچ اختلالی روانی ندارند و در 20 یا 30 سالگی به سر می برند؛ گزارش های گاه به گاه افراد 50 تا 60 ساله وجود دارد. از آنجا که اجرای درمان جایگزینی آنزیم برای سندرم هانتر وجود دارد ، انتظار می رود طول عمر افراد بدون ناراحتی روانی طولانی شود زیرا بیماری فیزیکی آنها با چنین درمانی بهبود یا تثبیت می شود.
کیفیت زندگی در تعداد زیادی از مردم همچنان بالا است و بسیاری از بزرگسالان به طور فعال مشغول به کار هستند.
در مقابل، افراد مبتلا به سندرم هانتر اختلالات روانی شدید، امید به زندگی 15 سال یا کمتر را دارند، که اغلب به علت عصبی شدن عصب یا عوارض جسمی بیماری است. سن شروع تشنج و وجود / عدم اختلالات رفتاری عامل پیش بینی کننده در بیماری بیماران بسیار جوان است. اختلالات رفتاری اغلب می تواند ترکیبی از علائم اختلال بیش فعالی کمبود توجه ، اوتیسم ، اختلال وسواس فکری و / یا اختلال پردازش حسی را تقلید کند ، هرچند وجود و میزان علائم در هر کودک آسیب دیده متفاوت است. آنها اغلب شامل فقدان حس مناسب خطر و تجاوز نیز می شوند.

پاتوفیزیولوژی
سندرم هانتر یا موکپوپلی ساکاریدوز 2 (MPS II) یک اختلال ژنتیکی جدی است که عمدتا در مردان ( X-linked recessive ) تأثیر می گذارد. این تداخل با توانایی بدن برای شکستن و بازیافت موکوپلی ساکارید خاص است که همچنین به عنوان گلیکواسامینو گلیکان یا GAG شناخته می شود. سندرم هانتر یکی از چندین بیماری ذخیره سازی لیزوزومی است که به نام بیماری های MPS شناخته می شود.
در سندرم Hunter، GAG به علت کمبود یا عدم وجود آنزیمی iduronate-2-sulfatase (I2S) در سلولهای بدن ایجاد می شود. این ساخت و ساز باعث می شود که سلول ها و اندام های خاص بدن متاثر گردند و منجر به تعدادی از علائم جدی شود. همانطور که افزایش GAG در سراسر سلول های بدن ادامه می یابد، نشانه های سندرم هانتر بیشتر قابل مشاهده است.نشانه های فیزیکی برخی از افراد مبتلا به سندرم هانتر شامل ویژگی های چهره مشخص و سر بزرگ است. در بعضی موارد سندرم هانتر، درگیری سیستم مرکز عصبی منجر به تاخیر رشد و مشکلات سیستم عصبی می شود. همه افرادی که مبتلا به سندرم هانتر هستند دقیقا همان شیوه بیماری را تحت تاثیر قرار می دهند و میزان پیشرفت علامت ها به طور وسیعی متفاوت است. با این حال، سندرم Hunter همیشه شدید، پیشرفته و باعث محدودیت هایی در زندگی است، حتی زمانی که به عنوان "خفیف" یا "ملایم" شناخته شده است.
ژنتیک
از آنجایی که سندروم هانتر یک اختلال ارثی است ( که مبتنی بر X است ) که به طور عمده در مردان تاثیر می گذارد، از یک نسل به بعد به روش خاصی منتقل می شود. تقریبا هر سلول در بدن انسان 46 کروموزوم دارد که 23 مورد از هر یک از والدین حاصل می شود. ژن IDS بر روی کروموزوم X واقع شده است. زنان دارای دو کروموزوم X هستند که از هر پدر و مادر به ارث می رسد، در حالی که مردان دارای یک کروموزوم X هستند که از مادرشان و یک کروموزوم y که از پدر آنها به ارث برده می شوند.
اگر یک مرد نسخه ی غیر طبیعی از ژن IDS داشته باشد، او سندرم Hunter را توسعه خواهد داد. یک مرد می تواند یک نسخه ی غیر طبیعی از ژن IDS را در یکی از دو روش بدست آورد. مادرش اغلب حامل است؛ یعنی او دارای یک ژن غیر طبیعی و یک ژن طبیعی IDS است و او ژن غیر طبیعی را از او میگیرد.
حالت دوم :متناوبا، در طی تخمک گذاری و شکل گیری اسپرم، جهش در ژن IDS در کروموزوم X خود رشد ایجاد می شود. در مورد دوم، مادر حامل نیست و خطر ابتلا به جهش خود به خودی دوباره در آینده خواهر و برادر کم است، اما نه صفر است. خانم ها می توانند یک نسخه ی غیر طبیعی از ژن IDS را حمل کنند و معمولا تحت تاثیر قرار نمی گیرند.
منظر بیوشیمی
بدن انسان به یک مجموعه گسترده ای از واکنش های بیوشیمیایی برای حمایت از عملکرد های حیاتی، از جمله تولید انرژی ، رشد و توسعه ، ارتباط درون بدن و حفاظت از عفونت وابسته است. یکی دیگر از ویژگی های مهم تجزیه بیومولکول های بزرگ است که مشکل اساسی در سندرم هانتر (MPS II) و اختلالات مربوط به ذخیره سازی است.
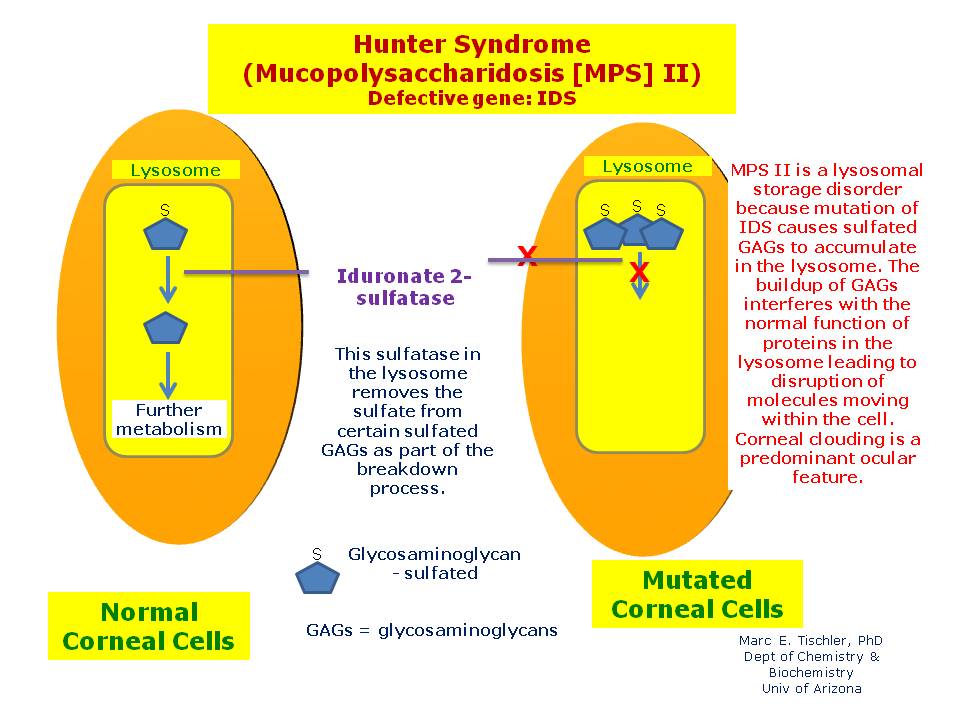
بیوشیمی سندرم هانتر به یک مشکل در بخشی از بافت همبند بدن شناخته شده به عنوان ماتریکس خارج سلولی مربوط می شود . این ماتریس از انواع قندها و پروتئین تشکیل شده و به تشکیل چارچوب معماری بدن کمک می کند. ماتریس سلول های بدن را در یک شبکه مشبک سازماندهی می کند و به عنوان چسب عمل می کند که سلول های بدن را با هم نگه می دارد. یکی از قسمتهای ماتریکس خارج سلولی یک مولکول پیچیده است که پروتئگلیکان نامیده می شود. پروتئگلیکان ها مانند بسیاری از اجزای بدن باید شکسته شوند و جایگزین شوند. هنگامی که بدن پروتئگلیکان ها را از بین می برد، یکی از محصولات حاصل از آن موکو پلی ساکارید است که در غلظت های گلیکوآمینوگلیکان (GAG) نامیده می شود. انواع مختلفی از GAG وجود دارد که هر کدام از آنها در مکان های خاصی از بدن یافت می شوند.
در سندرم هانتر، این مشکل مربوط به تجزیه دو GAG: سولفات درماتان و سولفات هپاران است . اولین گام در شکست سولفات درماتان و سولفات هپاران نیاز به آنزیم لیزوزوم I2S دارد. در افراد مبتلا به سندرم هانتر، این آنزیم یا بخشی از آن و یا کاملا غیر فعال است. در نتیجه، GAG در سلول ها در سراسر بدن ایجاد می شود، به ویژه در بافت هایی که حاوی مقادیر زیادی از درماتان سولفات و هپاران سولفات است. همانطور که این ساخت و ساز ادامه می یابد، از آن جا که این تجمع ادامه می یابد تداخل با مسیر های واکنش سلولی، منجر به تعدادی از نشانه های جدی می شود. میزان تولید GAG برای همه افراد مبتلا به سندرم هانتر یکسان نیست و طیف وسیعی از مشکلات پزشکی را به همراه دارد.
تشخیص
علائم و نشانه های سندرم Hunter (MPS II) در افراد جوان معمولا اولین سرنخ هایی هستند که منجر به تشخیص می شوند . به طور کلی، زمان تشخیص معمولا در حدود 2 تا 4 سالگی اتفاق می افتد. پزشکان می توانند از طریق آزمایش های آزمایشگاهی برای اندازه گیری فعالیت آنزیم ایپورونات 2-سولفاتاز (I2S)، قبل از تشخیص قطعی، شواهد بیشتری ارائه دهند که اختلال MPS وجود داشته باشد. آزمایش غربالگری اغلب برای یک اختلال MPS ، یک آزمایش ادرار برای GAG است. مهم است که توجه داشته باشیم که آزمایش ادرار برای GAG ها گاهی اوقات طبیعی می باشد و در عین حال هنوز کودک ممکن است اختلال MPS داشته باشد. تشخیص قطعی سندرم Hunter با اندازه گیری فعالیت I2S در سرم ، گلبول های سفید خون یا فیبروبلاست ها که بیوپسی پوست ساخته می شود امکان پذیر است . در برخی از افراد مبتلا به سندرم هانتر، تجزیه و تحلیل ژن I2S می تواند شدت بالینی را تعیین کند. تشخیص قبل از زایمان به طور معمول توسط اندازه گیری فعالیت آنزیمی I2S در مایع آمنیوتیک یا بافت ویولون کوریونی قابل دسترس است.
درمان
به دلیل ماهیت بسیار خاص بیماری ، درمان بسیار دشوار است. درمان برای این اختلال به طور خاص برای هر بیمار مشخص می شود، زیرا همه موارد متفاوت هستند.
درمان تسکین دهنده
با توجه به ماهیت بیماری و فقدان یک درمان واقعا کارآمد، مهم است که توجه به درمان گسترده ای در برابر علائم مختلف داشته باشیم. هدف آنها کاهش اثرات زوال بسیاری از اعمال بدن است. با توجه به تنوع علائم، معمولا از طیف گسترده ای از استراتژی های تسکین دهنده استفاده می شود که جراحی و درمان اغلب محوری هستند.
پیوند مغز استخوان
برای مدت زمان طولانی، کارآمد ترین روش استفاده از پیوند مغز استخوان یا پیوند سلول های بنیادی خونی بود . آنها هر یک از مزایای ارائه منبع جدید از I2S گم شده را دارند. با این حال، نتایج به بهترین شکل در نظر گرفته ناقص است.
در حالی که این جایگزین درمان می تواند پیشرفت برخی علائم فیزیکی را بهبود بخشد یا متوقف کند، پسرفت شناختی ناگهانی که در بیماران مبتلا به سندرم هانتر رخ می دهد، شناخته شده نیست، اگرچه ممکن است این پسرفت را در اوایل کند کند. بنابراین، برای بیماران مبتلا به کمبود، هنوز هم ممکن است به عنوان یک گزینه درمان مناسب، به دلیل ماهیت دائمی آن استفاده شود..
با این حال، حتی برای بیماران مبتلا به آن، مداخله خطرناکی است که خطر مرگ و میر قابل توجهی دارد و ممکن است عواقب خطرناکی یا تغییرناپذیری مانند بیماری پروتئین در برابر میزبان داشته باشد. به علت همه این دلایل، پیوند مغز استخوان یا پیوند سلول های بنیادی خونی، مصرف دارو را به عنوان درمان سندرم هانتر کاهش می دهد.
جایگزینی آنزیم
Idursulfase ، یک فرم خالص از آنزیم لوسوسومی iduronate-2-sulfatase تولید شده توسط تکنولوژی DNA نوترکیب در یک سلول انسانی ، در سال 2006 تحت آزمایش بالینی قرار گرفت و متعاقبا توسط اداره غذا و داروی ایالات متحده به عنوان درمان جایگزینی آنزیم برای سندرم هانتر تصویب شد
اپیدمیولوژی
تخمین زده می شود تقریبا 2000 نفر مبتلا به سندرم هانتر در سراسر جهان، 500 نفر از آنها در ایالات متحده زندگی می کنند. 2 بیمار مبتلا به سندرم هانتر در نیوزیلند، 6 بیمار مبتلا به سندرم هانتر در ایرلند، حداقل 1 مورد در ایران ، 1 مورد در عربستان سعودی، 1 مورد در شیلی، 1 مورد در پاکستان، 20 مورد در فیلیپین، 1 مورد در کرانه غربی (فلسطین) و 70 بیمار مبتلا به سندرم هانتر در کره گزارش دادند .
تاریخچه
این سندرم به دلیل زحمات پزشک چارلز هانتر (1873-1955) نامیده می شود، که اولین بار در سال 1917 آن را توصیف کرد. متولد اسکاتلند، هانتر به کانادا مهاجرت کرد و در وینیپگ، مانیتوبا، یک کار پزشکی داشت.
 آشنایی با شاخه های مختلف زیست شناسی
آشنایی با شاخه های مختلف زیست شناسی